Infectious Disease/Disease Non-Battle Injury
The WPAC PMO develops safe and effective therapeutics, vaccines, diagnostics and assays for all relevant threats. We develop vaccines and prophylaxis critical for protecting deployed Service Members against existing and emerging diseases that degrade unit readiness. Deployable diagnostics enable rapid pathogen detection to quickly identify and diagnose infectious diseases in the field enhancing far-forward medical response where evacuation times may be extended. These broad-spectrum capabilities mitigate infections, reduce morbidity, improve recovery and enable the Warfighter's return to the fight.
Adenovirus Type 4 and 7 Vaccine
Adenovirus Vaccine is used exclusively by the military to prevent Adenovirus-related acute respiratory disease in Soldiers and other military trainees living in barrack-type environments during basic training. The vaccine consists of two orally administered, enteric-coated tablets, with one containing live adenovirus serotype 4 and the other containing type 7 adenovirus. Prior to the use of Adenovirus Vaccines, adenovirus types 4 and 7 accounted for at least 60 percent of all acute respiratory disease in military recruits who were hospitalized.
Adenoviruses are associated with pharyngitis, conjunctivitis, atypical pneumonia and rhinitis. The development effort for the Adenovirus Vaccine was conducted primarily under a contract to Barr Laboratories, Inc. (now a subsidiary of Teva Pharmaceutical Industries Ltd.). The FDA approved the Biologics License Application in March 2011 and vaccine deliveries started in October 2011. Continued surveillance at the training sites shows that the Adenovirus Vaccine remains highly effective. It prevents an estimated 15,000 cases of acute respiratory disease each year, and thereby helps the DOD avoid the loss of approximately 50,000 training days. Over two million doses of the vaccine have been shipped to the immunization sites to date. The Defense Logistics Agency, in coordination with the WPAC PMO, awarded the Full-Rate Production contract in February 2020, to supply the military requirements of the Adenovirus vaccine through 2029. The WPAC PMO responsibilities include central management, central funding, serving as the requisitioning agency and distribution activity for the vaccine. Additionally, a contract was awarded to TEVA in the fourth quarter of fiscal year 2020 to conduct a gap analysis to review the current production methods for the vaccine with a focus toward improving the manufacturing process to ensure long-term viability of the product.
BioFire Defense COVID-19 Test 510(k) and Sample Type Expansion (CARES Act)
FDA-cleared in vitro diagnostic device used with BioFire FilmArray® for qualitative detection of nucleic acid from SARS-CoV-2 in nasopharyngeal swabs (NPS) in viral transport media. Sample type expansion studies to support amendment of the existing Emergency Use Authorization (EUA).
The DOD owns hundreds of FilmArray instruments. It has used that testing platform to deliver COVID-19 testing capabilities to Military Health System beneficiaries. USAMMDA WPAC manages the advancement of the regulatory status of BioFire Defense's COVID-19 test, which has been used under EUA since March 2020. The studies done to support FDA clearance by the 510(k) route will provide additional performance information for the test and assure that its use is not limited to the duration of the COVID-19 emergency. USAMMDA WPAC also manages the effort to extend the utility of the COVID-19 test, currently limited to nasopharyngeal swabs in transport media, to nasal and oropharyngeal swabs, sputum, and saliva, and to media that are more readily sourced than viral transport media.
In June 2020, a USAMMDA-led team awarded a contract to BioFire Defense for development of the COVID-19 test to FDA clearance, and in October 2020 awarded a separate contract for sample type expansion of the EUA test. In March 2021, the FDA authorized expansion of sample types to nasal and oropharyngeal swabs and sputum. Studies to support further expansion to saliva samples were in progress as of April 2021.
Broad Spectrum Snakebite Antidote (BSSA)
The BSSA is intended to be an FDA-approved, shelf-stable, broad-spectrum treatment for snakebite envenomation, designed to be easily used in austere conditions worldwide, regardless of snake species. A primary objective is to halt or reverse toxicity, thus widening the window of time allowed for safe evacuation and bite treatment.
Envenomation is a considerable risk to Warfighters operating in austere environments and there is no treatment readily available at Role 1 (i.e. point-of-injury or battalion aid station). Even when transport to higher echelons of care is possible, the only existing treatment available is administration of antivenom, if available. Antivenom is specific to snake species, carries a risk of adverse side effects (anaphylaxis, serum sickness), exists in limited quantities worldwide, and presents logistical and supportability challenges for far-forward administration. Therefore, a broad spectrum solution that can be easily used by a victim or a combat medic is desired. Our industry partner, Ophirex, Inc., is working to develop varespladib, a small molecule drug that inhibits a common component of snake venom and can stave off or reverse the toxic effects of envenomation. Prior clinical trials have shown varespladib to be safe in humans and pre-clinical data suggests efficacy against venoms from greater than 50 different snakes. The drug is shelf stable under tropical ambient conditions and can be administered orally in tablet form.
A research and development contract was awarded to Ophirex, Inc. in June 2020 for manufacturing and regulatory activities leading to a pivotal clinical trial that will test varespladib against envenomation in humans. Both oral (tablet) and IV (lyophilized) material suitable for a pivotal Phase 2b clinical trial was manufactured and is under an ongoing stability program. Clinical testing of the oral form of varespladib against bites from any species of snake will begin in late spring 2021 in the U.S. and India. The Materiel Development Decision/Milestone (MS) A brief was conducted on February 19, 2020, and entered into the Technology Maturation and Risk Reduction (TMRR) phase.
ARDS PLA2 Inhibitor for SARS-CoV2 Treatment (CARES Act)
Varespladib is a small molecule PLA2 inhibitor originally under development as a broad-spectrum snakebite antidote (see BSSA) and now currently under a repurposing development effort for COVID-19 associated-ARDS treatment. Through increased levels of a protein called phospholipase A2 (PLA2), COVID-19 can cause a cytokine storm and disruption of lung surface that results in increased inflammation and lack of oxygen distribution to organs. This is collectively known as Acute Respiratory Distress Syndrome (ARDS) and is a leading cause of death due to COVID-19. As with BSSA, varespladib can be given orally (tablet) or IV to patients developing or experiencing ARDS. The objective of the ARDS PLA2 inhibitor for SARS-CoV2 treatment is to provide a treatment option that will mitigate the symptoms of COVID-19 that lead to death and severe respiratory issues. This solution is not an antiviral and is therefore expected to work independent of coronavirus strain, and may have utility against other causes of ARDS. As a repurposing effort, this program is intended to move quickly to clinical trial evaluation.
A research and development contract was awarded to Ophirex, Inc. in June 2020. Ophirex manufactured both oral and IV clinical trial material and prepared a clinical protocol. The company anticipates clinical trial initiation in late spring/summer 2021.
Dengue Countermeasures: Dengue Tetravalent Vaccine (DTV)
The Dengue Tetravalent Vaccine is for the prevention of infection caused by any of the four serotypes of the dengue fever virus. The Dengue virus has four different serotypes and is the most significant arthropod-borne viral disease of man, and the most geographically widespread. The distribution of dengue virus has increased 30-fold in the past 50 years. Annually, it is estimated that there are nearly 100 million symptomatic infections, of which 500,000 cases require hospitalization, leading to 10,000 deaths. Dengue is epidemic or endemic in over 128 countries, primarily in the tropic and sub-tropic regions where military personnel are/or will be stationed or deployed. Impacted areas include Western Pacific, Southeast Asia, Central and South America, Caribbean and Africa. Increasingly, the U.S. and its territories are impacted, to include: Puerto Rico epidemic outbreaks in 2010 and 2012; parts of Florida now considered endemic; and sporadic outbreaks in Hawaii, including the 2015/2016 outbreak during which more than 264 people were infected. The National Center for Medical Intelligence (NCMI) sets incidence rates at greater than 10 percent per month in parts of Southern Command (e.g., Colombia) and Pacific Command (e.g., Thailand). Dengue epidemics are explosive, with high attack rates and morbidity, and with the potential to rapidly incapacitate large numbers of personnel.
The illness caused by dengue virus is characterized by sudden onset of fever, severe headache, pain behind the eyes, generalized joint and muscle aches, lack of appetite, gastrointestinal disturbances and rash. In about five percent of the cases, the infection can progress to debilitating and sometimes fatal manifestations of the disease referred to as Dengue Hemorrhagic Fever (DHF) and Dengue Shock Syndrome. Mortality for unmanaged DHF may reach 30 percent. In U.S. troops, it is estimated that each case of dengue will lead to 14 lost-duty days. The NCMI estimates that a force of 100,000 would endure 2,000 cases per month in areas where attack rates exceed 10 percent. Currently, there is no vaccine or drug to prevent the disease, that has U.S. Military utility, and treatment consists primarily of supportive care with extensive hospitalization and the potential for mortality.
As part of the Cooperative Research and Development Agreement (CRADA) between the USAMRDC and the industry partner, the MRDC's leading DTV candidate is currently in Phase 3 clinical studies, in the Philippines at the Armed Forces Research Institute of Medical Sciences, which is the Walter Reed Army Institute of Research (WRAIR) OCONUS laboratory in Thailand. This live, attenuated vaccine is administered in two doses, 90 days apart.
The vaccine candidate is being tested for efficacy and safety; the primary efficacy endpoint data was published in November 2019. In March 2020, the industry partner published the secondary endpoint data reporting the 18-month efficacy and 48-month safety data. The industry partner intends on submitting for licensure to the FDA in the second half of calendar year 2021. The goal is to achieve FDA approval within 12 months after submission.
Enterotoxigenic E. coli Vaccine (ETECV)
The ETECV is intended to be an FDA-approved multivalent vaccine indicated for active immunization of adults against enterotoxigenic E. coli to prevent bacterial diarrhea. Prevention of diarrhea would curtail a considerable source of lost duty days, particularly in the first month of deployment, while minimizing antibiotic treatment in theater and the possibility of long-term gastrointestinal issues. Infectious diarrhea is repeatedly the most common illness reported by travelers visiting low- and middle-income countries abroad, and among U.S. troops deployed overseas. The impact of bacterial diarrhea on unit readiness caused it to be ranked the number one infectious disease threat by the Combatant Commands Infectious Diseases Threat Prioritization Panel (through the Military Infectious Disease Research Program) in 2019. Approximately 80 percent of infectious diarrhea cases are caused by bacterial agents, of which ETEC causes the greatest proportion. Although the military has developed extensive capabilities for the provision of clean food and water, diarrhea has continued to be a problem for deployed personnel. The ETECV is intended to prevent ETEC-attributable diarrhea using a multivalent strategy to achieve strain coverage.
A research and development contract was awarded to Scandinavian Biopharma on September 25, 2020, for manufacturing of clinical trial material suitable for a Phase 3 clinical trial. Scandinavian Biopharma is developing the lead candidate, a whole cell killed vaccine comprised of 6 different immunogenic components. Technical batches of vaccine have been produced at-scale and are undergoing testing. Phase 2 trials tested the vaccine in adult European travelers to Benin. The MDD/MS A brief was conducted on April 27, 2020, and entered into the TMRR phase.
Human Immunodeficiency Virus Vaccine (HIVV)
The HIVV will provide a globally effective FDA-licensed vaccine for the prevention of infection by HIV-1 to Service Members. Following identification of Human Immunodeficiency Virus (HIV) and acquired immunodeficiency syndrome (AIDS) in the U.S. population, the U.S. Congress initiated the U.S. Military HIV Research Program in 1986 to reduce the impact of HIV in the military through prevention, treatment, diagnostics and monitoring. HIV infection and the resulting AIDS illness pose a threat to the individual readiness of U.S. personnel. Service Members must be prepared to deploy globally and potentially on short notice; due to policy, however, HIV-positive active duty personnel are not deployable, with limited exceptions.
Globally, an estimated 37.9 million people are infected with HIV, and 1.7 million new infections were reported in 2018. The latest data from the Medical Surveillance Monthly Report (MSMR, August 2019) indicates that, despite implementation of HIV prevention education programs and bi-annual testing of Service Members, the number of new infections has remained constant from 2011 to 2019, from 300 to 350 per year.) HIV is spread by sexual contact with an infected person, by sharing needles and/or syringes (primarily injection drug users), or less commonly, through transfusions of infected blood or blood products.
HIV causes AIDS, which is a severe syndrome characterized by progressive damage to the immune system and other organs, including the central nervous system. Currently, there is no cure for the infection, only lifetime drug therapy to limit viremia and prevent advancement to the severe form of AIDS. Personnel infected while serving are provided lifetime care through the DOD and VA healthcare systems, which is estimated to cost $1.29 million for therapeutic drugs alone over the expected lifetime of Service Members post-infection, which is 40 to 50 years.
HIV epidemiology is complex, with 10 subtypes (A, B, C, D, F, G, H, J, K and L) and more than 35 circulating recombinant forms (CRF). The main subtypes of interest for vaccine development are A, B, C, D, and CRF01_AE; the major subtype in the U.S. is B, which represents 96 percent of DOD and general population infections. There is no vaccine available to prevent infection and disease caused by HIV.
The current vaccine supported by the WPAC PMO was developed jointly over the past 10 years by Janssen Vaccines and Prevention B.V. (Janssen), the U.S. Military, Beth Israel Deaconess Medical Center and the National Institutes of Health, National Institute for Allergy and Infectious Diseases, Division of AIDS (DAIDS). The Janssen vaccine is a prime-boost regimen consisting of an adenovirus 26 (Ad26) vector with mosaic gene inserts for gag and pol, or env sequences (prime component) and two glycoprotein (gp) 140 env proteins (boost component). The Ad26 mosaic inserts and gp140 proteins were designed using bioinformatics to select sequences by in silico recombination, and have been optimized to elicit cellular and humoral responses against multiple circulating subtypes HIV-1. USAMMDA signed a CRADA with Janssen Vaccines and Prevention B.V. (Janssen) to support Phase 2 and Phase 3 clinical studies in April 2017, and awarded an Other Transaction Agreement (OTA) in 2019 to help fund the Phase 3 efficacy trial. The Army is one of several collaborators supporting Janssen in their development efforts, and include the DAIDS, the HIV Vaccine Trials Network, the Bill and Melinda Gates Foundation, and the Ragon Institute at Harvard University. The Phase 2b Proof of Concept efficacy study was initiated in November 2017, and the study is testing the vaccine regimen in five southern African countries, including Malawi, Mozambique, South Africa, Zambia and Zimbabwe. This ongoing study focuses on healthy women who are at risk of HIV infection, to test the efficacy of the regimen in a region where infections are primarily from HIV- 1 subtype C. Janssen formally announced the start of its Phase 3 efficacy study, named MOSAICO in July 2019. This study focuses on high-risk individuals (men who have sex with men and transgender women) and its intent is to assess efficacy of the vaccine against subtype B infections. The Phase 3 trial will be conducted in North America, Europe and Latin America.
The Phase 3 study initiation and Army collaboration was announced at a key international AIDS conference in July 2019. The first site in the U.S. was activated the end of October 2019, in Houston, Texas. Results from the Phase 2b study are expected in 2021, and from the Phase 3 study in 2024.
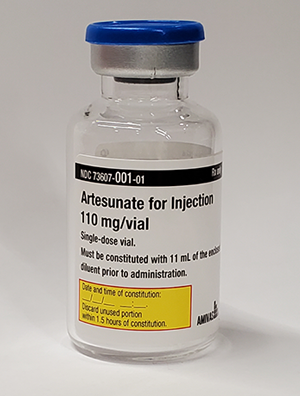
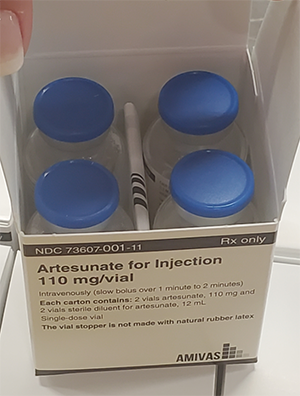
Malaria Treatment Drug – Intravenous Artesunate (MTD-IVAS)
MTD-IVAS provides an FDA-approved drug for the initial treatment of Service Members afflicted with severe and complicated Plasmodium falciparum malaria. The need for MTD-IVAS is critical in that malaria remains one of the most important infectious disease threats to Service Members during deployment to tropical and subtropical areas, and malaria has a long history as the cause of disease and non-battle injury to military personnel. The drug achieved FDA approval on May 26, 2020. The USAMRDC assisted in the New Drug Application (NDA) approval by providing chemistry, manufacturing and controls and clinical subject matter expertise. Amivas, LLC holds the FDA licensure and will manufacture and distribute the commercial product in fiscal year 2021. The FHP treatment Investigational New Drug (IND) protocol will continue to be used for treatment of cases of severe malaria until the commercial product becomes available.
Artemisinin, the naturally isolated parent drug of artesunate, is extracted from "qing hao" or sweet wormwood (Artemisia annua L.). It has been part of traditional Chinese herbal medicine for centuries. Artesunate was rediscovered and isolated as the active antimalarial agent in Artemisia annua L. in 1972 by Chinese scientists, and later independently by scientists at WRAIR seeking new treatments for malaria. Several million malarial patients have been treated with artesunate produced in China, and it has been found to be highly effective in rapid parasite clearance and fever reduction when given by the oral, intramuscular or IV route. Almost every publication refers to it as being a safe, effective and predictable drug for the treatment of severe and complicated malaria. A landmark study in Southeast Asia published in 2005 found that MTD-IVAS has a 35 percent all-cause mortality benefit over quinine for the treatment of severe malaria. This study showed that MTD-IVAS is more effective and superior to quinine, the more than 300-year-old gold standard for the treatment of malaria.


Since 1991, quinidine has been the only parenterally administered drug available in the United States for the treatment of severe malaria, but it is not an ideal drug. Quinidine is associated with sudden cardiac death, principally via cardiac arrhythmias, and, because of its short half-life, must be administered two to three times a day in an intensive care setting. Most significantly, quinidine is no longer the drug of choice in treating certain electrocardiac disturbances. Since March 2019, quinidine is no longer available in the U.S. With the discontinued availability of IV quinidine, MTD-IVAS is the only FDA approved drug in the U.S for the treatment of severe malaria.
MS C was achieved on March 2, 2021, and will proceed to enter the Operations and Support (O&S) phase once Full Operation Capability (FOC) has been achieved.
Malaria Prophylactic Drug – Tafenoquine (MPD-TQ)
The standard of care for the prevention of malaria includes personal protection measures (vector control, repellents, permethrin-treated uniforms and bed nets) and the administration of FDA-approved malaria prophylactic drugs, which currently includes mefloquine, doxycycline, chloroquine and atovaquone/proguanil (Malarone™). Personal protection measures reduce the risk of malaria, but have limited efficacy in operational settings. Additionally, the currently available malaria chemoprophylaxis drugs have limitations that include dosing regimen (daily vs. weekly), tolerance and side effects, drug resistance and cost.
Tafenoquine (TQ) is an 8-aminoquinolone antimalarial drug first synthesized at WRAIR in 1978. This class of drugs has activity against all stages of malaria parasites, and several clinical studies have shown that TQ is very effective in preventing Plasmodium (P.) falciparum and P. vivax malaria in endemic areas. Furthermore, TQ can prevent relapsing malaria caused by P. vivax and P. ovale. Concurrent with the Army's development effort, GlaxoSmithKline (GSK), through a former licensing agreement with the Army, has developed TQ for the radical cure of P. vivax indication.
The use of TQ requires prescreening for normal glucose-6-phosphate dehydrogenase (G6PD) levels, which is routinely performed on all U.S. Service Members upon entry into service. G6PD deficiency is a hereditary condition, and the use of TQ in such individuals can result in hemolytic anemia.
An alternative anti-malarial drug with a weekly dosing regimen for the prevention of malaria will be developed, procured, fielded and administered to deployed military and support personnel. MPD-TQ is immediately anticipated to replace mefloquine, which retains a black box warning from the FDA but was the only weekly prophylactic drug available prior to TQ approval. Then, gradual replacement of daily chemoprophylaxis with MPD-TQ is expected to improve overall compliance, reduce pill burden on users, and reduce burden of direct observed therapy on leadership, together leading to reduced need for hospitalization and evacuation as well as savings in the costs of associated medical care in high-transmission areas.

60° Pharmaceuticals, LLC (60P), the USAMRDC corporate partner for the development of TQ for prophylaxis, submitted an NDA package to the FDA for TQ for Malaria Prophylaxis, and it was granted approval on August 8, 2018, with the trade name Arakoda™. GSK received FDA approval for TQ for Radical Cure of P. vivax malaria (trade name Krintafel™) on July 20, 2018. Both indications for TQ were also approved by the Australian Therapeutic Goods Administration in 2018. Post-marketing commitments for the drug are currently in progress. Enrollment for a clinical study to assess long-term safety of Tafenoquine was completed on February 7, 2020, and the study is expected to close out in 2021. This study is also evaluating expanded indication to 12 months of continual use. On March 3, 2020, Arakoda™ was added to the Joint Deployment Formulary.
Next Generation Diagnostics System Increment 1 Device – Infectious Disease Panel (NGDS-IDP)

The NGDS Increment 1 device is a FDA–cleared, ruggedized, reusable, adaptable diagnostic device for the analysis of biological (clinical) liquid samples. Its diagnostic capabilities provide health care providers with timely and accurate information to inform individual patient treatment. This effort focuses on developing an ID-GFP kit that can be used with samples of whole blood in conjunction with this device.
Infectious diseases typically result in seven to 14 lost-duty days, prolonged troop performance impacts that may last years, and even, in some cases, death. Historically, for deployed personnel, infectious disease rates occur at 100 cases per 1,000 troops.
The Infectious Disease Global Fever Panel (ID-GFP) kit, to be used with the Next Generation Diagnostics System Increment 1 (NGDS) device, provides the capability to detect in a multiplexed assay format as many as 17 fever-causing pathogens to include parasites, viruses and bacteria. The NGDS device and ID- GFP kit will be utilized in Army ROC 3, Air Force ROC 2 to 3 and Navy ROC 2 to 3 deployable Combat Health Support units to provide sensitive, specific, and rapid (within 70 minutes) detection of nucleic acids from these fever-causing pathogens, enabling faster and better informed medical decisions. This system supports near-real-time patient treatment and health protection decision-making.
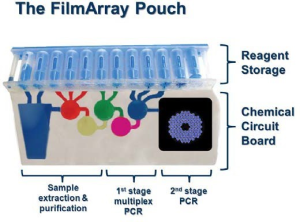
The sponsor of the NGDS ID-GFP kit is BioFire Defense, LLC. This effort is a collaboration between Joint Project Manager for Chemical, Biological, Radiological, and Nuclear, Medical (JPM CBRN Medical), National Institute of Allergy and Infectious Diseases, BioFire Defense LLC (contractor), and USAMMDA, with each partner responsible for funding the development of required assays for their diseases of interest. The ID-GFP kit underwent extensive testing with blood samples obtained in clinical studies performed in the U.S., South America, Africa, and Asia. In 2020, BioFire Defense submitted applications to the FDA for approval of the ID-GFP tests for Chikungunya, Dengue, Leptospirosis, and Malaria and an External Control Kit to be used by laboratories to validate their performance of the tests. The FDA approved those applications in November 2020, setting the stage for deployment in July 2021.
Rapid Human Diagnostics (RHD)
The Tropical Fever (TF) panel is an FDA-cleared rapid diagnostic assay in development for diagnosis of military-relevant infectious diseases. The TF panel is a consumable assay cartridge packaged to include all ancillary components (i.e. pipette) required to run the assay on a hand-held, battery-powered device used to analyze clinical specimens. The TF panel will facilitate early diagnosis of dengue, chikungunya, zika, malaria, leptospirosis, and Salmonella typhii to inform medical treatment and intervention decisions, ultimately returning the Warfighter to combat.
Tropical diseases can cause significant impact to force readiness and combat effectiveness, spreading quickly through units, causing a substantial number of lost duty days, and potentially incapacitating large numbers of Service Members during conflict. Early detection plays an important role in the mitigation of infection, especially during field deployments where full laboratory facilities may be limited or unavailable. Therefore, having laboratory capabilities that can extend far-forward to ROC 1 and 2 and in austere conditions where diseases are often encountered first is important. Diagnostics play a valuable and critical role in patient care by clarifying the etiology of the illness and informs treatment decisions; reduces the risk of patients unknowingly transmitting the disease to others, ultimately, preventing or stopping an outbreak.
On September 23, 2020, USAMMDA's WPAC PMO entered into a Memorandum of Agreement (MOA) with the Joint Program Manager (JPM) Chemical, Biological, Radiological and Nuclear (CBRN) Medical for the co-development of the TF panel. Together, the Army and JPM CBRN Medical will manage the development of the TF panel through the EMD acquisition phase of the Next Generation Diagnostic System Increment 2 (NGDS-2)/Man-Portable Diagnostic System (MPDS) program through MS C and fielding. The NGDS-2/MPDS program achieved MS B in September 2019, leveraging a U.S. Special Operations Command (USSOCOM)-validated Capability Development Document (CDD). Approved in March 2020, a Joint CDD aligns testing strategies and fielding approaches for the NGDS-2/MPDS program among all Services.
The partnership with JPM CBRN Medical is a shift in acquisition strategy for the RHD program from single to multi-target diagnostic assays. The goal is to deliver a significantly enhanced diagnostic capability more quickly than possible under the Army's previous acquisition strategy and at a significantly reduced cost. As of March 2021, the NGDS-2/MPDS program is preparing to initiate clinical studies to assess TF panel performance and conduct an operational assessment among a far-forward small mission team. The MGDS-2/MPDS program MS C/LRIP decision will occur as early as the first quarter of fiscal year 2022, which will initiate production of final-form systems of the NGDS-2/MPDS device. According to the JRO CDD, the Army is last among all of the Services to field the NGDS-2/MPDS device and TF panel, which will occur as early as March 2024.
The NGDS-2/MPDS device will be available via a follow-on procurement contract with the developer for Army specific units, in Medical Companies, at ROC 1 to 2 and incorporated into the Lightweight Medical Equipment/Materiel Sets. The TF panel will be available to the Combatant Commands for procurement, based on mission need, using the Common Table of Allowances (CTA) 8-100 program through a Pharmaceutical Prime Vendor contract.
The JPEO CBRND Medical MPDS program MS C brief will occur in the first quarter of fiscal year 2022 (O)/third quarter of fiscal year 2022 (T). The TF panel is scheduled to achieve FDA-clearance as early as the fourth quarter of fiscal year 2022.
SCoV-2 Rapid Diagnostic Lateral Flow Tests (CARES Act)
The novel coronavirus, SARS-CoV-2 (the causative agent of COVID-19), has been responsible for the pandemic of pneumonia-like symptoms and associated deaths from late 2019 through 2020 globally. The detection of the initial outbreak in the Hubei Province of China and the subsequent need for high-performing and rapid diagnostics were quickly recognized. The World Health Organization declared SARS-CoV-2 a global pandemic on March 11, 2020. Soon after, the President of the United States declared a National Emergency on March 13, 2020.
The presence of SARS-CoV-2 virus is detectable in respiratory specimens from subjects displaying signs and symptoms of acute SARS-CoV-2 infections, including cough, fever, headache, muscle pain, chills, sore throat, and respiratory distress.
It has been reported the polymerase chain reaction (PCR) confirmed patient positive specimens may seroconvert and develop antibodies against SARS-CoV-2 antigens anywhere from 6-21 days after the onset of clinical symptoms. The specific and reliable detection of human IgM antibodies to SARS-CoV-2 may provide early detection of infection within days after the onset of clinical symptoms. The specific and reliable detection of human IgG antibodies to SARS-CoV-2 remains a key method to monitor infections, to effect contact tracing and for serosurveillance.
In a whole-of-government response to the COVID-19 pandemic, the U.S. government through the Defense Health Agency and CARES Act funding made investments in a broad range of medical products (i.e. diagnostics, vaccines, and treatments). One of the diagnostic efforts was an FDA-authorized (i.e. EUA) rapid diagnostic assay for diagnosis of SARS-CoV-2 in symptomatic and asymptomatic patient from a nasopharyngeal swab (from individuals who are suspected of COVID-19 by their healthcare provider) for direct antigen (SCoV-2 Ag Detect™) detection and whole blood, plasma, or serum serology (SCoV-2 Ab Detect™) detection. Both the direct antigen detection and serology tests take less than 30 minutes to obtain results, making a suitable diagnostic tool for use in a point-of-care setting.
InBios funded early assay development for both SCoV-2 Ab Detect™ and SCoV-2 Ag Detect™. BARDA funded SCoV-2 Ab Detect™ development activities to achieve EUA. DHP CARES Act funding also supported SCoV-2 Ag Detect™ development activities to achieve EUA. The FDA granted EUA to InBios on May 6, 2021 for its qualitative SCoV-2 Detect antigen rapid test in direct anterior nasal swab specimens from individuals suspected of having COVID-19.
(January/February 2021). USAMMDA's WPAC PMO has partnered with BARDA for future development of these rapid tests, and both groups plan on entering into an Interagency Agreement to facilitate BARDA funding development activities in FY21-22 to achieve 510(k) clearance for all assays.